
Buy Semaglutide Research Peptide Online: A peptide for weight loss
Buy Semaglutide Research Peptide Online: A peptide
Quick Answer
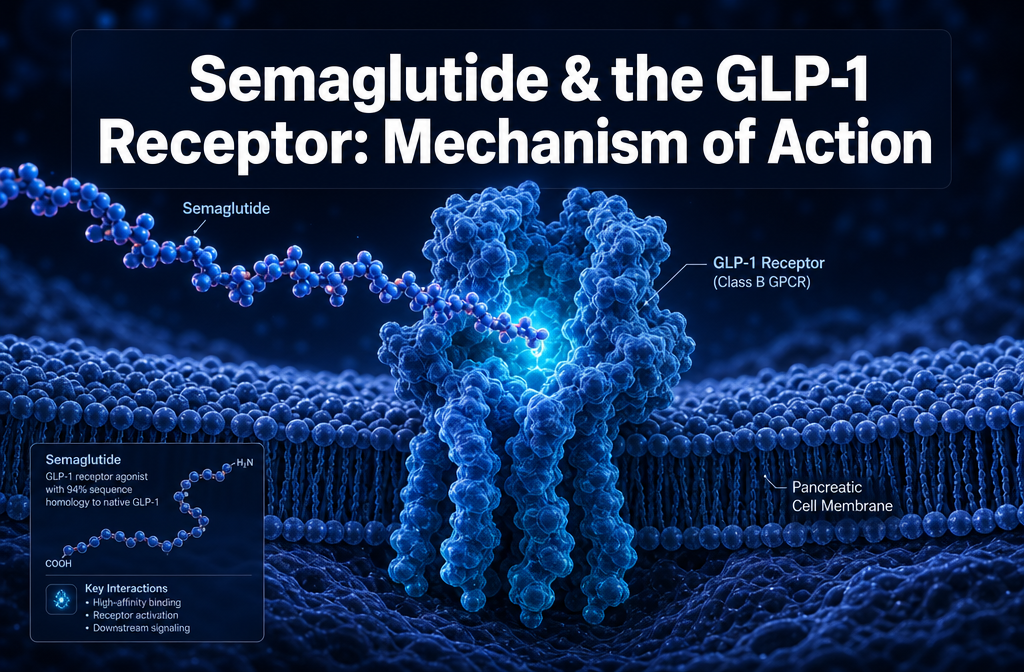
Semaglutide is a synthetic GLP-1 receptor agonist that works by binding to and activating glucagon-like peptide-1 (GLP-1) receptors across the pancreas, gut, and central nervous system. This activation stimulates glucose-dependent insulin secretion, suppresses glucagon release, slows gastric emptying, and reduces appetite signalling in the hypothalamus. Its structural modifications give it a half-life of approximately seven days, making it far more durable than native GLP-1, which is degraded within minutes.
Semaglutide is a 34-amino acid synthetic peptide classified as a GLP-1 receptor agonist. It is structurally derived from native human GLP-1 but engineered with specific modifications that dramatically extend its biological activity. In preclinical and clinical research, it functions by mimicking the action of endogenous GLP-1 at its target receptors, producing a coordinated set of metabolic effects that include enhanced insulin secretion, glucagon suppression, decelerated gastric emptying, and reduced appetite signalling [8].
The compound was developed to overcome the primary limitation of native GLP-1: a plasma half-life of approximately two minutes, caused by rapid degradation by the enzyme dipeptidyl peptidase-4 (DPP-4) and renal clearance. Semaglutide addresses this through acylation with a C18 fatty diacid moiety linked via a hydrophilic spacer, which promotes tight binding to serum albumin. This albumin association shields the peptide from DPP-4 and reduces renal filtration, extending the half-life to roughly seven days [1].
For researchers working across metabolic and endocrine research areas, semaglutide represents one of the most well-characterised GLP-1 pathway tools available. A detailed overview of its pharmacological profile is available in our guide on what semaglutide is and how it works.
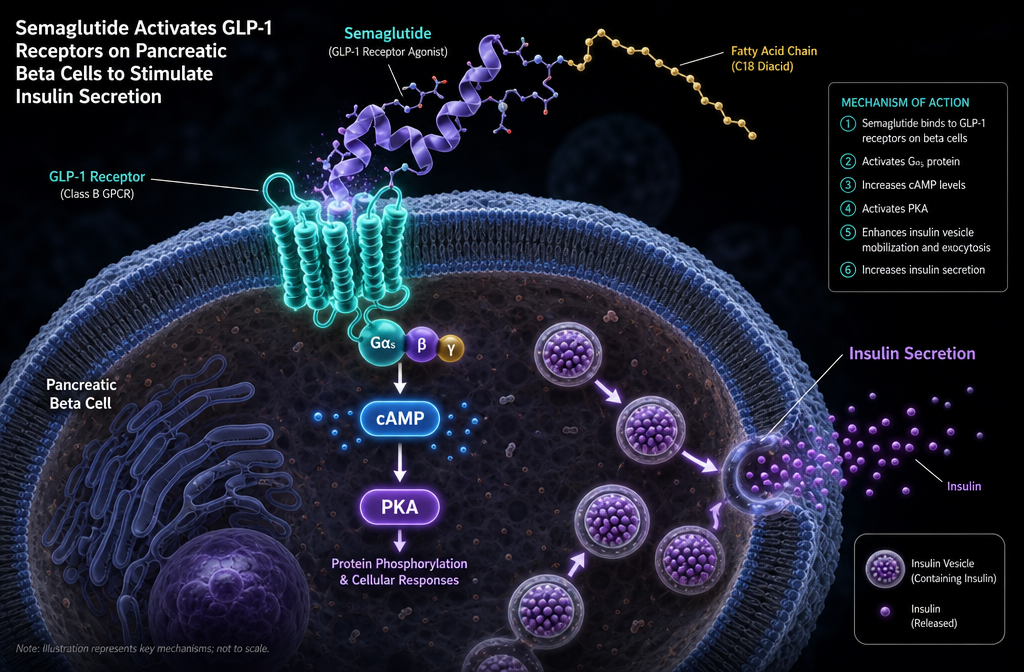
The GLP-1 receptor (GLP-1R) is a class B G protein-coupled receptor (GPCR) expressed in multiple tissues, including pancreatic beta cells, the gastrointestinal tract, the hypothalamus, the hindbrain, the heart, and the kidneys. When activated, it couples primarily to the Gs protein, triggering adenylyl cyclase to produce cAMP, which then activates PKA and exchange proteins directly activated by cAMP (EPACs). This signalling cascade is the central mechanism through which semaglutide produces its metabolic effects [4].
The receptor’s distribution across multiple organ systems is precisely why the semaglutide mechanism of action GLP-1 receptor interaction produces such a broad physiological response. Pancreatic GLP-1R activation drives insulin secretion; hypothalamic GLP-1R activation suppresses appetite; and GLP-1R activation in the gut slows motility. Each of these effects is initiated by the same receptor-binding event but produces tissue-specific outcomes depending on the local cellular machinery.
Understanding GLP-1R biology is foundational for researchers designing metabolic studies, because the receptor’s expression level, coupling efficiency, and downstream signalling capacity can all vary across tissue types, experimental models, and genetic backgrounds.
Semaglutide mimics native GLP-1 through structural homology and receptor selectivity. It shares 94% sequence homology with human GLP-1(7-37), meaning the amino acid sequence responsible for receptor recognition is largely preserved [1]. The two key structural differences — a substitution at position 8 (alanine replaced by aminoisobutyric acid to resist DPP-4 cleavage) and the addition of the C18 fatty acid chain at position 26 — do not disrupt receptor binding. Instead, they extend the compound’s plasma residence time while maintaining full agonist activity.
Where semaglutide surpasses the native peptide is in potency. Research data indicate an EC50 of approximately 6.2 pM for semaglutide at the GLP-1 receptor, compared to 16.2 pM for native GLP-1 — representing approximately 2.6-fold greater receptor activation efficiency [7]. This enhanced potency is attributed to the structural modifications that allow the peptide to adopt a conformation that engages the receptor binding domain more effectively.
In practical terms for laboratory research, this means that semaglutide activates the same downstream signalling pathways as endogenous GLP-1 — cAMP production, PKA activation, and EPAC-mediated effects — but does so with greater efficiency and over a substantially longer timeframe.
Semaglutide’s effects on blood glucose operate through three coordinated mechanisms, all initiated by the semaglutide mechanism of action GLP-1 receptor binding event.
Glucose-dependent insulin secretion: When semaglutide binds to GLP-1 receptors on pancreatic beta cells, it elevates intracellular cAMP, which activates PKA. PKA enhances insulin gene transcription and promotes the exocytosis of insulin-containing secretory granules. Critically, this process is glucose-dependent — it is amplified only when blood glucose is elevated, which is why GLP-1 receptor agonism carries a low intrinsic risk of hypoglycaemia when used as a monotherapy [4].
Glucagon suppression: Semaglutide also acts on GLP-1 receptors expressed on pancreatic alpha cells, suppressing glucagon secretion in a glucose-dependent manner. Since glucagon drives hepatic glucose production, its suppression reduces fasting and postprandial glucose output from the liver [5].
Delayed gastric emptying: GLP-1 receptors in the gastrointestinal tract mediate a slowing of gastric motility. Semaglutide activates these receptors, delaying the rate at which ingested nutrients enter the small intestine and subsequently appear in circulation. This blunts postprandial glucose excursions without altering fasting glucose to the same degree [5].
Native GLP-1 has a plasma half-life of approximately two minutes. Semaglutide has a half-life of approximately seven days. This difference — roughly 5,000-fold — is the defining pharmacokinetic distinction between the endogenous peptide and its synthetic analogue [1].
This extended duration results from three structural features working in combination. First, the substitution at position 8 blocks DPP-4-mediated degradation at the primary cleavage site. Second, the C18 fatty acid chain promotes reversible, high-affinity binding to serum albumin, effectively creating a circulating reservoir that slowly releases free peptide. Third, albumin binding reduces glomerular filtration, decreasing renal clearance significantly.
For the oral formulation, absorption dynamics differ. Peak plasma concentrations are typically reached within one hour of oral administration, with steady-state exposure achieved after four to five weeks of daily dosing [3]. This is relevant for researchers comparing subcutaneous and oral administration routes in metabolic models.
Semaglutide reduces body weight primarily through central nervous system mechanisms, acting on GLP-1 receptors expressed in the hypothalamus and hindbrain. Hypothalamic GLP-1R activation suppresses appetite by modulating the activity of pro-opiomelanocortin (POMC) neurons and inhibiting neuropeptide Y (NPY)/AgRP neurons, both of which regulate energy intake [6].
Research published in Nature Metabolism indicates that semaglutide induces weight loss through cAMP-dependent mechanisms specifically in GLP-1 receptor-expressing neurons in the hindbrain, suggesting a direct central neural pathway that operates independently of peripheral metabolic effects [2]. This finding has significant implications for researchers investigating the neurological basis of appetite regulation and energy homeostasis.
The combined effect of reduced appetite signalling, delayed gastric emptying (which prolongs satiety), and improved insulin sensitivity creates a multi-pathway reduction in caloric intake and metabolic dysregulation. Researchers exploring body composition models should note that these effects are mechanistically distinct from direct lipolytic activity — semaglutide reduces energy intake rather than directly mobilising stored fat.
For comparison with compounds that activate additional receptor pathways, the tirzepatide research compound adds GIP receptor agonism, while retatrutide activates GLP-1, GIP, and glucagon receptors simultaneously, producing additive effects on energy expenditure.
The GLP-1 receptor agonist class includes several compounds with meaningful pharmacological differences. The table below summarises the key distinctions relevant to research applications.
| Compound | Half-Life | Receptor Targets | Dosing Frequency | EC50 at GLP-1R |
|---|---|---|---|---|
| Native GLP-1 | ~2 minutes | GLP-1R only | Continuous infusion | ~16.2 pM |
| Exenatide | ~2.4 hours | GLP-1R only | Twice daily | Higher than semaglutide |
| Liraglutide | ~13 hours | GLP-1R only | Once daily | Moderate |
| Semaglutide | ~7 days | GLP-1R only | Once weekly | ~6.2 pM [7] |
| Tirzepatide | ~5 days | GLP-1R + GIPR | Once weekly | Dual agonism |
| Retatrutide | ~6 days | GLP-1R + GIPR + GcgR | Once weekly | Triple agonism |
Semaglutide’s primary advantage over earlier GLP-1 agonists is its combination of extended half-life and superior receptor potency. Compared to tirzepatide and retatrutide, it offers a more targeted GLP-1R-specific pharmacological profile, which is valuable for research designs that require isolation of GLP-1 pathway effects without confounding GIP or glucagon receptor activity. A detailed comparison of multi-pathway incretin compounds is available in our GLP-1/GIP multi-pathway metabolic research guide.
In preclinical research, semaglutide is primarily used in models investigating type 2 diabetes, obesity, cardiovascular disease, and increasingly, neurological conditions including cognitive decline. Its well-characterised GLP-1 receptor mechanism makes it a reference compound for metabolic pathway research [8].
Genetic factors can influence how effectively semaglutide activates downstream signalling. Variants in the GLP1R gene affect receptor expression levels, binding affinity, and G protein coupling efficiency. Research in human populations has identified single nucleotide polymorphisms (SNPs) in GLP1R that are associated with differential glycaemic responses to GLP-1 receptor agonists. Subjects with reduced GLP-1R expression or impaired receptor coupling may show attenuated responses to semaglutide, which is a relevant variable in research model selection and data interpretation.
Additionally, if a research model presents with substantially reduced GLP-1R density — whether due to genetic variation, disease-related receptor downregulation, or tissue-specific expression differences — the magnitude of semaglutide’s effect on insulin secretion and appetite suppression may be diminished. Researchers should account for receptor expression levels when designing comparative studies or interpreting inter-subject variability in outcomes.
Semaglutide carries a low intrinsic risk of hypoglycaemia when used as a monotherapy because its insulin-stimulating effect is glucose-dependent. The cAMP-PKA signalling cascade in beta cells is only amplified when intracellular glucose metabolism is active, meaning that at normal or low blood glucose concentrations, the incremental insulin release driven by GLP-1R activation is minimal [4]. Hypoglycaemia risk increases when semaglutide is combined with insulin or sulphonylureas in clinical settings, as those agents operate through glucose-independent mechanisms.
Regarding missed doses in research protocols: semaglutide’s seven-day half-life means that a single missed weekly administration does not produce an abrupt loss of pharmacological effect. Plasma concentrations decline gradually, and the compound’s albumin-bound reservoir continues to release active peptide over several days. In research models using weekly subcutaneous administration, a missed dose will reduce steady-state exposure incrementally rather than causing an immediate return to baseline receptor activity.
The most frequently observed adverse effects in research models and clinical studies are gastrointestinal in nature and are mechanistically linked to the semaglutide mechanism of action GLP-1 receptor activation in the gut. These include nausea, vomiting, diarrhoea, and constipation. The underlying cause is the compound’s effect on gastrointestinal motility — specifically the slowing of gastric emptying mediated by GLP-1 receptors in the enteric nervous system and gut wall [5].
These effects are typically most pronounced at the initiation of dosing and during dose escalation, as GLP-1 receptors in the gut are exposed to higher concentrations than they would encounter with endogenous GLP-1. They tend to attenuate as receptor adaptation occurs over several weeks.
Researchers should also note that central GLP-1R activation in the area postrema (a circumventricular organ involved in emetic signalling) contributes to nausea. This is a direct consequence of the same CNS mechanism that suppresses appetite, as the area postrema expresses high levels of GLP-1 receptors and responds to circulating semaglutide concentrations [2].
The semaglutide mechanism of action GLP-1 receptor interaction represents one of the most thoroughly characterised signalling pathways in modern metabolic research. By binding selectively to GLP-1 receptors across the pancreas, gastrointestinal tract, and central nervous system, semaglutide initiates a coordinated cAMP-PKA signalling cascade that drives glucose-dependent insulin secretion, suppresses glucagon, delays gastric emptying, and reduces appetite through both hypothalamic and hindbrain mechanisms.
Its structural modifications — the position-8 amino acid substitution and C18 fatty acid acylation — are not incidental. They are precisely engineered to overcome the limitations of native GLP-1, producing a compound with 2.6-fold greater receptor potency and a half-life approximately 5,000 times longer than the endogenous peptide.
For research professionals designing metabolic studies, the following steps are recommended:
Semaglutide remains the foundational reference compound for GLP-1 receptor research, and its mechanistic depth continues to generate productive lines of investigation across metabolic, cardiovascular, and neurological research domains.
Research Use Only Disclaimer: Products sold on this website are intended for research purposes only. They are not for human consumption, medical use, or therapeutic application. By purchasing from this website, you confirm that you are a qualified professional and will use these products strictly for laboratory research.
What is the primary mechanism by which semaglutide lowers blood glucose?
Semaglutide binds to GLP-1 receptors on pancreatic beta cells, elevating intracellular cAMP and activating PKA, which enhances insulin secretion in a glucose-dependent manner. It simultaneously suppresses glucagon from alpha cells, reducing hepatic glucose output [4].
Why does semaglutide have a much longer half-life than natural GLP-1?
Native GLP-1 is rapidly degraded by DPP-4 and cleared renally, giving it a half-life of about two minutes. Semaglutide’s position-8 amino acid modification blocks DPP-4 cleavage, and its C18 fatty acid chain promotes albumin binding, which reduces renal clearance and extends the half-life to approximately seven days [1].
Is semaglutide selective for GLP-1 receptors only?
Yes. Semaglutide is a selective GLP-1 receptor agonist and does not activate GIP or glucagon receptors at pharmacologically relevant concentrations. This distinguishes it from tirzepatide (dual GLP-1R/GIPR) and retatrutide (triple GLP-1R/GIPR/GcgR) [8].
How does semaglutide reduce appetite in research models?
Semaglutide activates GLP-1 receptors in the hypothalamus and hindbrain, modulating neuronal circuits that regulate energy intake. Research indicates that cAMP-dependent signalling in hindbrain GLP-1R-expressing neurons is a direct mechanism of appetite suppression [2].
Does semaglutide cause hypoglycaemia on its own?
The risk is low when used as a monotherapy because its insulin-stimulating effect is glucose-dependent — it amplifies insulin secretion only when blood glucose is elevated. Hypoglycaemia risk increases when combined with insulin or sulphonylureas [4].
What is the EC50 of semaglutide at the GLP-1 receptor compared to native GLP-1?
Semaglutide has an EC50 of approximately 6.2 pM at the GLP-1 receptor, compared to approximately 16.2 pM for native GLP-1, indicating roughly 2.6-fold greater receptor activation potency [7].
Why are gastrointestinal effects common with semaglutide?
GLP-1 receptors are expressed throughout the gastrointestinal tract and in the area postrema. Semaglutide’s activation of these receptors slows gastric motility and stimulates emetic signalling pathways, producing nausea, vomiting, and altered bowel habits, particularly during dose initiation [5].
Can genetic variation affect semaglutide’s effectiveness in research models?
Yes. Variants in the GLP1R gene can alter receptor expression levels and coupling efficiency. Research models with reduced GLP-1R density or impaired G protein coupling may show attenuated responses to semaglutide, which should be accounted for in experimental design.
What is the difference between subcutaneous and oral semaglutide in terms of pharmacokinetics?
Subcutaneous semaglutide achieves steady-state plasma concentrations over several weeks with once-weekly dosing. Oral semaglutide reaches peak plasma concentrations within approximately one hour of administration, with steady state achieved after four to five weeks of daily dosing [3].
How does semaglutide compare to tirzepatide for metabolic research?
Semaglutide acts exclusively on GLP-1 receptors, making it the preferred tool for GLP-1R-specific research. Tirzepatide adds GIP receptor agonism, producing additive effects on insulin secretion and body composition. Researchers should select based on whether pathway specificity or multi-receptor activation is required for their study design.
[1] Ozempic – https://www.drugs.com/pro/ozempic.html
[2] S42255 026 01534 8 – https://www.nature.com/articles/s42255-026-01534-8.pdf
[3] Mechanism Of Action (Rybelsus) – https://www.novomedlink.com/diabetes/products/treatments/rybelsus/about/mechanism-of-action.html
[4] PMC11790292 – https://pmc.ncbi.nlm.nih.gov/articles/PMC11790292/
[5] Mechanism Of Action (Ozempic) – https://www.novomedlink.com/diabetes/products/treatments/ozempic/about/mechanism-of-action.html
[6] Semaglutide – https://www.cancer.gov/publications/dictionaries/cancer-drug/def/semaglutide
[7] Semaglutide Research Overview – https://peptides.so/learn/semaglutide-research-overview
[8] NBK603723 – https://www.ncbi.nlm.nih.gov/books/NBK603723/
Buy Semaglutide Research Peptide Online: A peptide

Buy Tirzepatide Compound: The miracle peptide for

What is NAD+ and why does NAD+